文章来源:六月行研社
药苑杂谈 YAOYUANZATAN
药物临床试验期间、药品上市后都可以提出新增适应症的申请,本文总结了几种常见的新增适应症的情形,从法规依据和注册路径两方面概述,希望对大家有所启发和帮助,但可能存在考虑不周全的地方,欢迎各位同行指正。
情形一:临床试验期间,提出新增适应症
法规依据一:《药品注册管理办法》
法规依据二:《化学药品注册受理审查指南》(第一部分 注册分类1、2、5.1类)
对于新药,同一药物如有多个适应症,按照一个适应症对应一个IND/NDA提交注册申请;虽然受理审查指南对于仿制药(注册分类3、4、5.2类的产品)未作相关说明,但如果仿制的参比制剂有多个适应症,也应分别提交注册申请。
注册路径:IND+NDA/ANDA。
药品上市后新增适应症主要分为三种情况:
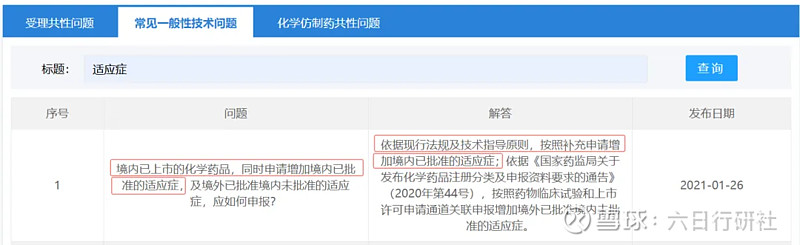
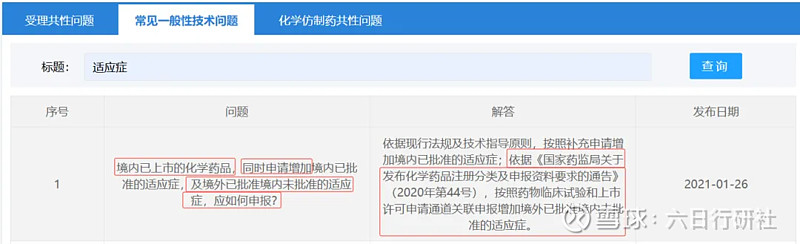
1)新增的适应症为境内已批准的,即国内同品种已批准的适应症(见情形二);
2)新增的适应症为境外已批准但境内未批准的,此处已上市药品包括仿制药和新药(分别见情形三、情形四);
3)新增的适应症为境内和境外均未获批的(情形五);
情形二:药品上市后,新增的适应症为境内
已批准的,即国内同品种已批准的适应症
法规依据一:《已上市化学药品和生物制品临床变更技术指导原则》
法规依据二:CDE常见一般性技术问题
根据法规要求,情形二属于重大变更,需要提交补充申请并经过审评、审批后执行。因新增的适应症为国内同品种已批准的,那么这个适应症的安全性和有效性是明确的。对于普通口服固体制剂,如果能做到药学一致、BE等效的话,大概率是不需要开展验证性临床。但如果是复杂制剂,比如复杂的注射剂,可能很难豁免,需具体问题具体分析。
注册路径:药品上市许可持有人在开展临床试验前,应首先提出补充申请,在获得批准后方可开展临床试验。药品上市许可持有人完成临床试验并经评估认为试验数据可支持相应变更时,可向国家药品监督管理局递交补充申请。
情形三:药品上市后(仿制药),
新增的适应症为境外已批准但境内未批准的
法规依据一:《化学药品注册受理审查指南》(第二部分 注册分类3、4、5.2类)
注册路径:IND+ANDA(3类)
情形四:药品上市后(新药),
法规依据:《化学药品注册受理审查指南》(第一部分 注册分类1、2、5.1类)
注册路径:IND+NDA(5.1类)
情形五:药品上市后,
新增的适应症为境内和境外均未获批的
注册路径:IND+NDA(2.4类)