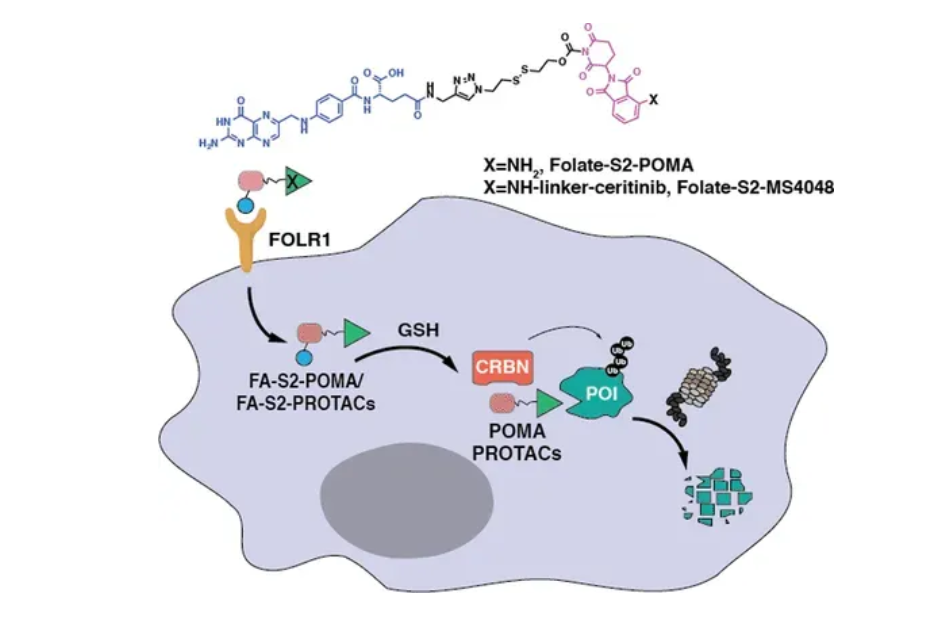
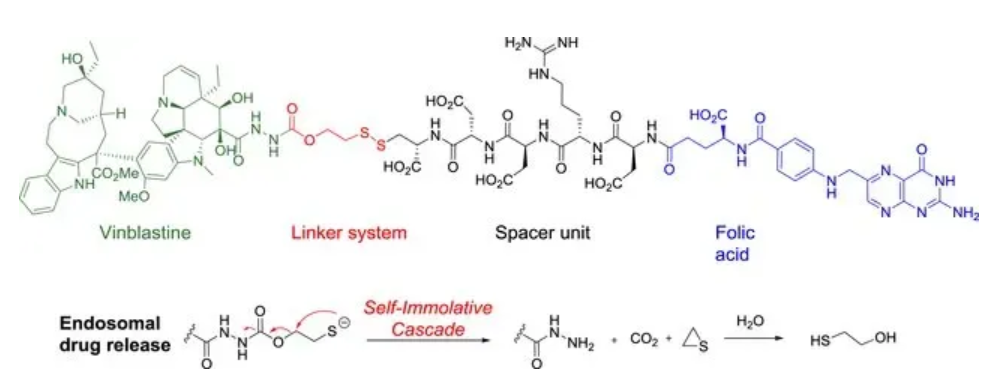
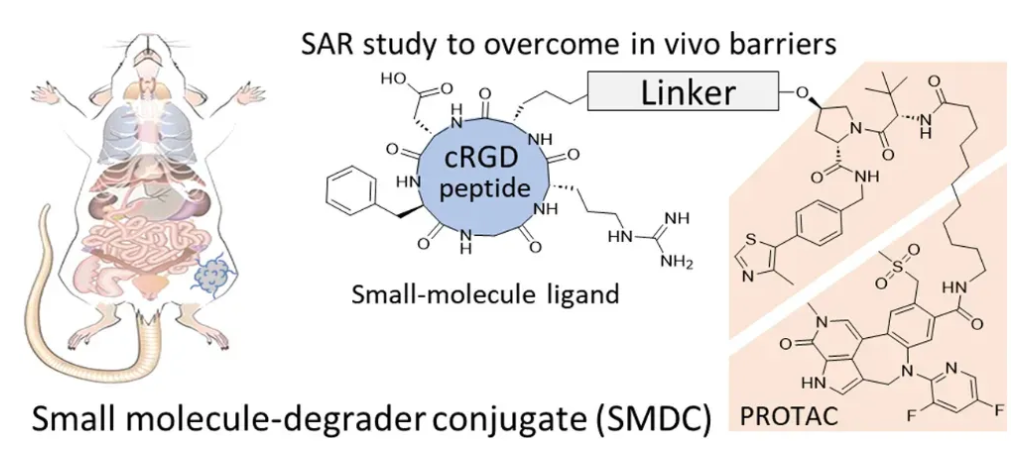
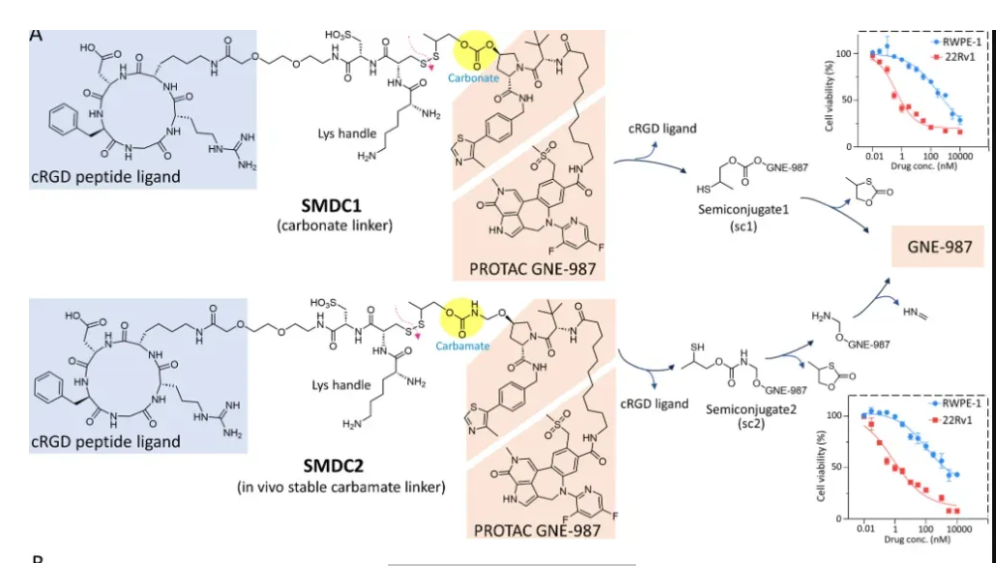
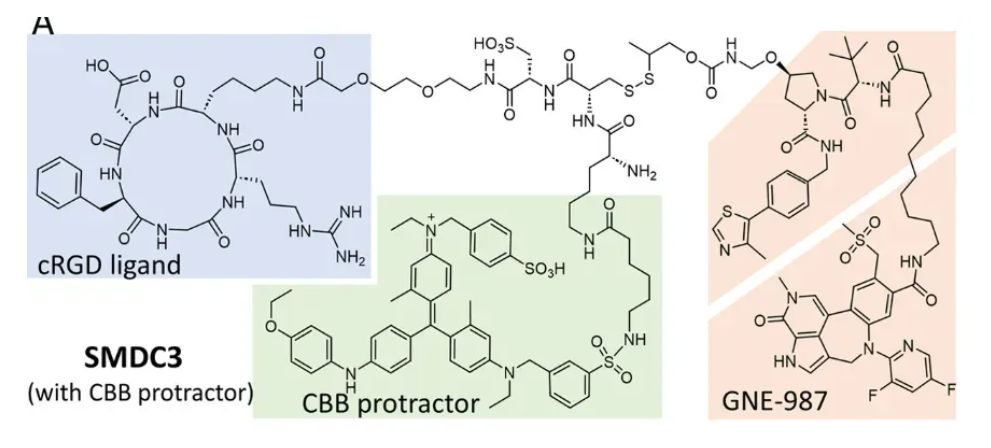
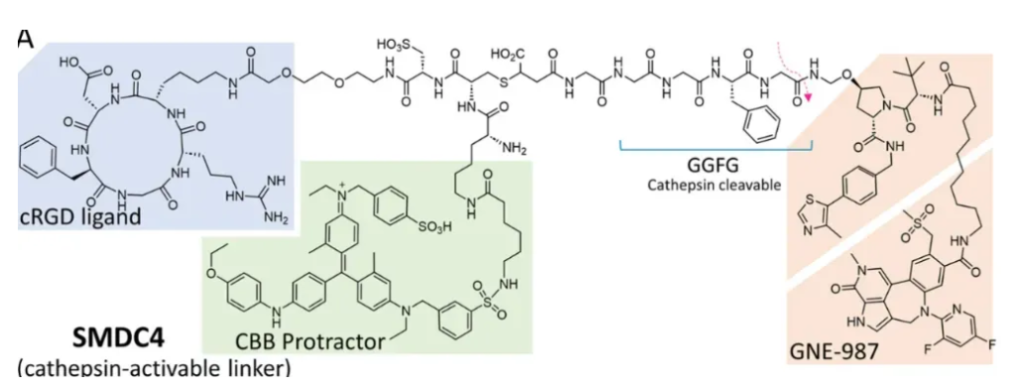
Small Molecule–Degrader Conjugates: Evaluating the Structure–Activity Relationship of Linkers to Overcome In Vivo Barriers in PROTAC Delivery, Shiwei Song, Weina Jing, Lei Peng, Jiaqi Liu, and Wanyi Tai. Journal of Medicinal Chemistry 2025 68 (16), 17323-17338. DOI: 10.1021/acs.jmedchem.5c00862
Chen, H.; Liu, J.; Kaniskan, H. Ü.; Wei, W.; Jin, J. Folate-Guided Protein Degradation by Immunomodulatory Imide Drug-Based Molecular Glues and Proteolysis Targeting Chimeras. J. Med.Chem. 2021, 64 (16), 12273– 12285, DOI: 10.1021
Dragovich, P. S.; Pillow, T. H.; Blake, R. A.; Sadowsky, J. D.; Adaligil, E.; Adhikari, P.; Bhakta, S.; Blaquiere, N.; Chen, J.; dela Cruz-Chuh, J.Antibody-Mediated Delivery of Chimeric BRD4 Degraders. Part 1: Exploration of Antibody Linker, Payload Loading, and Payload Molecular Properties. J. Med. Chem. 2021, 64 (5), 2534– 2575, DOI: 10.1021/acs.jmedchem.0c01845Miletić, N.; Weckesser, J.;Mosler, T.;Rathore, R.;Hoffmann, M. E.;Gehrtz, P.;Schlesiger, S.;Hartung, I. V.;Berner, N.;Wilhelm, S.Workflow for E3 Ligase Ligand Validation for PROTAC Development.ACS Chem. Biol.2025,20(2),507–521, DOI: 10.1021/acschembio.4c00812