
作为以全球药品审评监管程序完善著称且备受瞩目的监管机构,在过去的几年里,尤其是在疫情期间,FDA也正在遭受着前所未有的关注度以及巨大争议。近几年来,FDA有过紧急批准新冠疫苗以及新冠药物,对全球抗疫起到重要的高光时刻,也有过不顾业界质疑仍然宣布批准Biogen的阿尔兹海默病(AD)药物Aduhelm,让FDA公信力瞬间降到冰点的疑惑操作。
新年伊始,回顾2023年,作为疫情后的元年,FDA交出了怎样的答卷呢?答案是截止到圣诞节,FDA CDER(药品评价与研究中心,Center for Drug Evaluation and Research)目前已经批准了55款创新药,在数量上已经远超2022年的37款,和2020以及2021年全年批准的数量基本持平,而生物制品评价和研究中心(CBER)批准的基因疗法、细胞疗法、疫苗等数量也超过10款。
在2023年,FDA告别了在其任职超过四十年的传奇人物——珍妮特·伍德科克(Janet Woodcock),近40年来,她曾担任FDA代理局长、副局长和CDER主任等职务,宣告了一个时代的落幕。
在告别的同时,FDA在2023年也迎来了更多历史性事件。FDA宣布完全批准了继Aduhelm后真正意义上用于治疗阿尔兹海默病的药物Leqembi(lecanemab)、批准了来自大洋彼岸的中国PD-1药物Loqtorzi、批准了首款CRISPR基因编辑药物Casgevy上市……..在FDA已经批准的这55款创新药,就不乏上述充满历史意义的药物,这些药物将在美国甚至是全球范围内起到怎样的作用,又将怎样引领目前的全球医药市场,在未来几年都将会找到答案。
与此同时,根据报道2023年以来FDA累计发出了36份完整回复函(CRL),其中有近一半(47%)来自创新药NDA或者BLA申请,有6款新药在申报新适应症的过程中收到了FDA发出的CRL,此外,有5款改良型新药在申报新剂型、新规格的过程中收到了CRL,而这些药物,将有很大的可能宣布失败,背后的研发企业数十年的心血,也极大可能付之一炬。
有人欢喜有人愁,复杂的情绪夹杂,但这就是属于FDA,属于新药开发者,属于全球医药的2023年。
01 Leqembi仍饱受争议
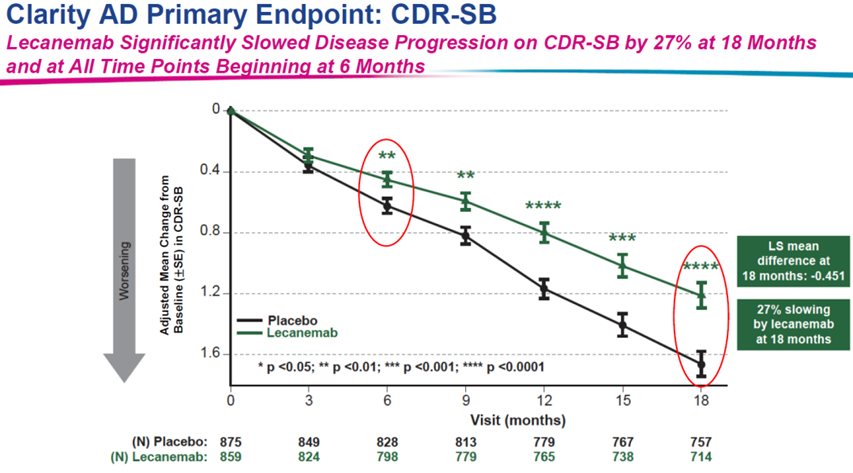
图1:Clarity AD临床试验结果(来自卫材官网)
02 RSV元年开启
03 中国PD-1出海成功
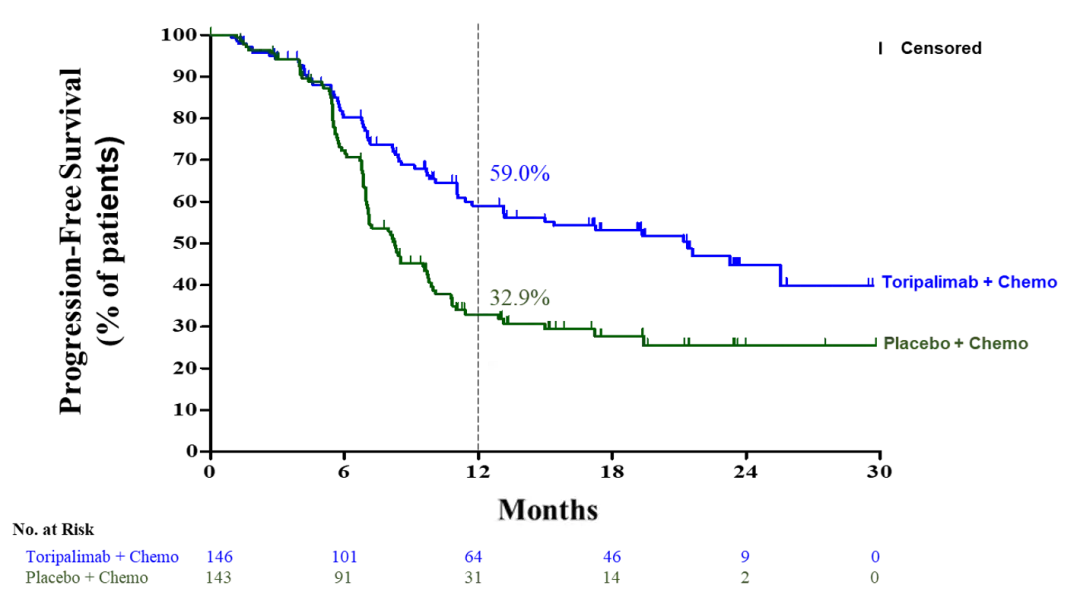
图2:JUPITER-02临床试验结果